Journal of Biomedical Engineering and Biosciences (JBEB)
ISSN: 2564-4998

Volume 10 - Year 2023 - Pages 36-40
DOI: 10.11159/jbeb.2023.006
Development of Sporobeads Coated With Hecad1/2 for Rapid Detection and Capturing Of Pathogenic Listeria Monocytogenes
Khosrow Mohammadi1, Per Erik Joakim Saris*1
1Department of Microbiology, Faculty of Agriculture and Forestry
University of Helsinki, P.O. Box 56, FI-00014 Helsinki, Finland
khosrow.mohammadi@helsinki.fi; per.saris@helsinki.fi
Abstract - Internalin proteins localized at the surface of pathogenic Listeria monocytogenes interfere with E-cadherin to adhere and internalize into mammalian cells. E-cadherin has five extracellular, immunoglobulin-like domains (EC1 to EC5), of which the first domain is sufficient to mediate L. monocytogenes invasion. Immunomagnetic beads employ antibodies that react either with pathogenic or non-pathogenic Listeria. To obtain cost-effective and easy-to-produce beads for detecting pathogenic L. monocytogenes, we cloned and efficiently expressed human E-cadherin domains 1 and 2 (hEcad1/2) in the Bacillus subtilis spore coat. The cDNA sequence encoding hEC1/2 protein 23.7 kDa (MH511517.1) was inserted in pET22b and expressed and purified from Escherichia coli BL21. We used the CotY as a significant structural component of the B. subtilis spore coat to express hEcad1/2. We constructed a recombinant plasmid p1CSV-CotY-N-hEcad1/2 incorporating the cotY-hEcad1/2 gene under the control of the cotY promoter. The constructed plasmid was transformed into B. subtilis KO7 by double cross-over method and an amylase-inactivated mutant was generated. After spore induction, the developed sporobeads showed a high binding affinity for L. monocytogenes 4b. This result demonstrated the potential of human E-cadherin ectodomain 1 and 2 in a practical application involving the detection and capture of pathogenic L. monocytogenes.
Keywords: Listeria monocytogenes, detection, E-cadherin, expression, Bacillus subtilis, sporobead.
© Copyright 2023 Authors This is an Open Access article published under the Creative Commons Attribution License terms. Unrestricted use, distribution, and reproduction in any medium are permitted, provided the original work is properly cited.
Date Received: 2023-08-20
Date Revised: 2023-09-25
Date Accepted: 2023-09-28
Date Published: 2023-10-16
1. Introduction
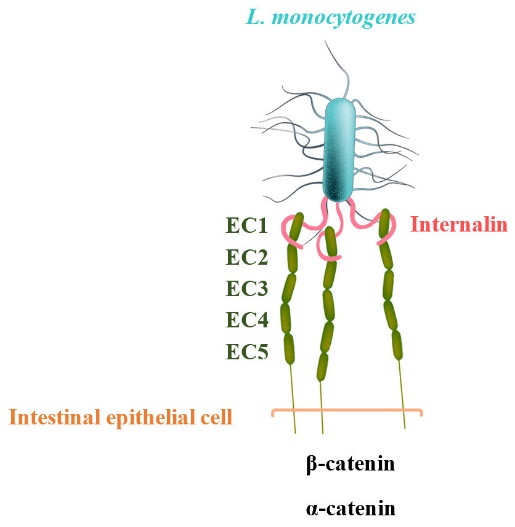
Listeria monocytogenes is one of the most important food-borne pathogens that causes a major opportunistic illness by crossing the intestinal epithelial barrier to access deeper tissues. In physiologically stressed and immunocompromised individuals, Listeria can cross the blood-brain barrier to spread to distant organs [1]. Adhesion and internalization of L. monocytogenes to the host intestinal epithelium are crucial for establishing infection and spreading to extraintestinal sites. In pathogenic Listeria species, internalin proteins (InlA, InlB, InlC, InlF, and InlP) control the stages of infection. The internalins InlA, InlB, and InlF are located at the bacterial cell surface and have a modular structure including an N-terminal cap domain, varying C-terminal repeats, an LRR-domain of 22 amino acid repeats, and an interrepeat region (IR) domain. These proteins target the adhesion molecule E-cadherin on the cell surface to bind and internalize into mammalian cells [2, 3].
E-cadherin has five extracellular, immunoglobulin-like domains (EC1 to EC5), of which the first domain is sufficient to mediate L. monocytogenes invasion. The leucine-rich repeat (LRR) domain of InlA surrounds and specifically recognizes hEC1. In other words, hEC1 occupies and fills the central cavity created by the curved LRR-domain of InIA (fig. 1) [2]. The pathogenicity of L. monocytogenes relies on virulence factors such as internalins [4]. In previous studies, bacteria expressing either InlA or InlB and beads coated with these proteins were successfully internalized in non-phagocytic cells [5]. In another related study, L. monocytogenes was shown to infect the E-cadherin-expressing cardiomyocytes in vitro [6].
Immunomagnetic separation (IMS) methods have been extensively used to detect L. monocytogenes. These methods employ antibodies that react with pathogenic or non-pathogenic Listeria [7]. The pathogenicity of L. monocytogenes relies on virulence factors such as internalins [8], and the hEcad ectodomain is adequate to start interaction with internalin, which connotes the cytoplasmic domain plays no role in adhesion [9].
Compared to the immobilization of proteins on synthetic surfaces, which is a costly and laborious procedure, the self-immobilization on B. subtilis spore coat has been introduced as a cost- and time-efficient alternative. Therefore, this study aimed to express human E-cadherin domains 1 and 2 (hEcad1/2) on the spore coat of B. subtilis and validate its potential in capturing pathogenic L. monocytogenes.
2. Materials and Methods
2.1. Construction of h-Ecad1/2 expression plasmid
The sequence coding for the extracellular domain of Human E-cadherin (657-bp hEcad1/2) (MH511517.1) was codon optimized and synthesized by Genescript and cloned into the pET22b with a 5′ overhang containing a XhoI site, a 3′ overhang containing NcoI site, and a 6× His-tag fused at the N-terminus of the expressed protein.
2.2. Expression of h-Ecad1/2
The expression plasmid pET22b-hEcad1/2 was cloned into competent E. coli BL21(DE3) cells by electroporation and positive transformed cells were selected on LB agar (merck) containing 100 μg/ml ampicillin. E. coli BL21 transformed with pET22b-hEcad1/2 was grown in 500 ml LB broth (merck) at 37 °C with vigorous shaking until OD 600 nm of 0.4-0.5 was reached, and an aliquot (t0) of supernatant was taken. Expression of the His-Tag- hEcad1/2 was induced by the addition of 1 mM isopropyl-1-thio-β-d-galactoside (IPTG) to a final concentration of 1 mmol/L, and one-milliliter sample was taken every hour, up to 5 h after induction. The cell pellet was harvested by centrifugation at 12,000 ×g for 5 min at 4 °C, resuspended in sonication buffer (50mM Tris-Hcl, 500mM NaCl) (pH 7.0), and sonicated at 4°C using a Sonorex Super 10P (Bandelin) ultrasonic bath. Cell debris was centrifuged at 12,000 ×g for 20 min at 4℃. The supernatant was prepared for the following purification.
2.3. Purification of hEcad1/2
The recombinant protein was purified using a HisTrap™ FF Crude column. The column was filled with Ni sepharose medium and equilibrated with binding buffer (20 mmol Na3PO4, 500 mmol NaCl, 5 mmol imidazole, pH 7.4). The sample was loaded at a flow rate of 0.5 ml/min, and the column was then washed with 10-15 column volumes of binding buffer. The column-bound proteins were eluted using elution buffer (20 mmol/L Na2HPO4, 1 mmol/L EDTA, and 1 mol/L NaCl, pH 8.0) at a flow rate of 1 ml/min. Protein fractions were analyzed by SDS-PAGE and stained with Coomassie Brilliant Blue.
2.4. SDS/PAGE and Western Blotting
The expression of target hEcad1/2 protein was analyzed using Bio-Rad mini-protean TGX precast gel on the PowerPac Basic (Bio-Rad, USA), electrophoresed at 100 V for 1 h, and then stained with Coomassie brilliant blue R250. Following SDS-PAGE, proteins were transferred from SDS-PAGE to polyvinylidene fluoride (PVDF) membranes on tank-blotting devices (Bio-Rad) at 400 mA for 60 min.
Western blot analysis was carried out using mouse anti-Histidine-Tag monoclonal primary antibody (Sigma) and alkaline phosphatase-conjugated goat anti-mouse secondary antibody (Sigma) and imaged by Bio-Rad ChemiDoc Touch Imaging system.
2.5. Construction of recombinant integration plasmid p1CSV-CotY-N-hEcad1/2 and pre-cloning in E. coli ECE371
The hEcad-specific primers, hec-F: 5′-TCTAGATAAGGAGGAAGCCGGCATGGACTGGGTTATTCCTCCCATCAG-3′ (XbaI site is underlined) and hec-R: 5′-ACTAGTATTAACCGGTCGGAGGATTATCGTTGGTGTCAG-3′ (NgoMIV site is underlined), were designed to amplify a 657-bp hEcad1/2 gene from the vector pET22b-hEcad1/2. The PCR product was purified using GeneJET PCR purification kit (Thermo Scientific) and then 5′-phosphorylated with T4 Polynucleotide Kinase (New England Biolabs) and linearized by T4 DNA ligase (Fermentas, USA). The purified PCR product (hEcad1/2 gene) was double digested with XbaI and AgeI while the p1CSV-CotY-N vector (rfp-cassette) was excised with XbaI and NgoMIV restriction enzymes and then fused to the end of the cotY gene to yield the p1CSV-CotY-N-hEcadc1/2 recombinant expression plasmid (Fig. 2).
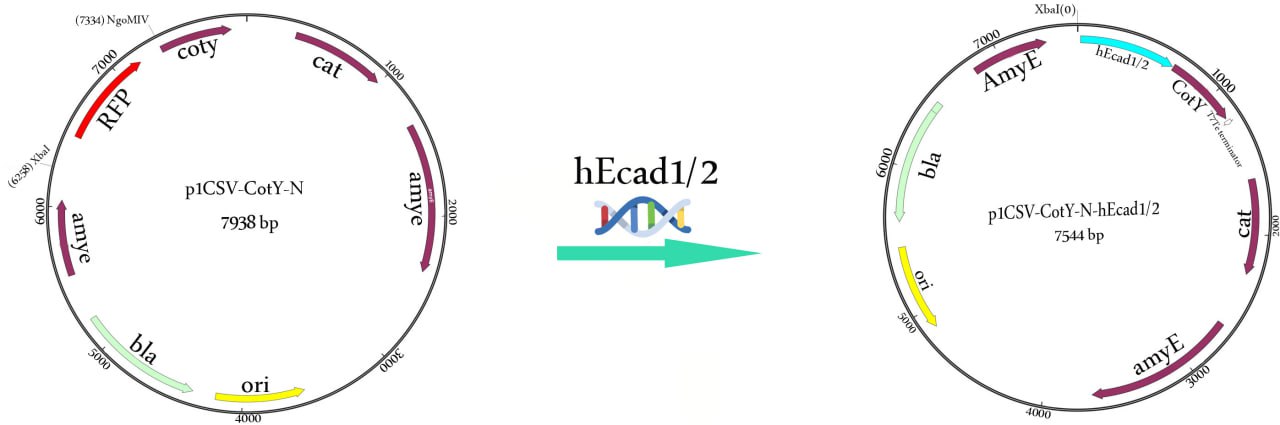
The vector contains a bla gene that mediates ampicillin resistance (100 μg/ml) in E. coli and chloramphenicol resistance (5 μg/ml) gene for selection in B. subtilis and integrates into the amyE locus by double crossing-over. For sequencing, E. coli ECE371 was transformed with p1CSV-CotY-N, and sequencing of the cloned hEcad gene was performed by Sanger sequencing.
2.6. Construction of recombinant Bacillus subtilis KO7
B. subtilis KO7 (Bacillus Genetic Stock Center ID 1A1133) was used to express the hEcad1/2 protein on the spore coat. The gene of hEcad1/2 was fused to the end of the cotY gene to yield the p1CSV-CotY-N-hec1/2 recombinant expression plasmid (Fig. 2). The integrated recombinant plasmid was linearized by ScaI and transformed into B. subtilis competent cells by double cross-over recombination with chloramphenicol selection (5 µg/ml). B. subtilis KO7 cells harboring p1CSV-CotY-N-hec1/2 were grown at 37 °C in LB medium containing chloramphenicol (5 µg/ml) to induce expression of the hEcad1/2 protein. The expected integration into the amyE locus was confirmed by the starch test, as described previously [10].
2.7. Preparation of spores
The exhaustion method was used to induce sporulation of B. subtilis in Difco Sporulation Medium (DSM), as previously described [11]. Cultures were harvested 48 h after incubation in DSM at 37 °C. Spores were collected, washed with 1 M NaCl, and purified using lysozyme treatment to break any residual sporulating cells.
2.8. Capture of L. monocytogenes
L. monocytogenes serotype 4b was activated and enriched in BHI broth at 37 °C for 24 h. After washing by 10 mM phosphate buffer solution (PBS) and centrifugation at 8000 rpm for 5 min, one hundred microliters of L. monocytogenes was added onto the ten microliters of spore, and the capturing efficiency was visualized under the microscope after a 10-min reaction.
3. Results
3.1. Verification of pET-22b containing hEcad1/2
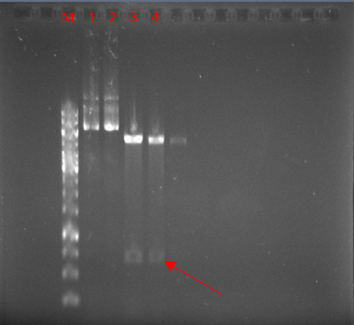
The vector was digested with NcoI and XhoI enzymes. As expected, a 671-bp band was observed after the digestion of pET22-hEcad1/2 (Fig. 3).
3.2. Protein expression and purification from E. coli
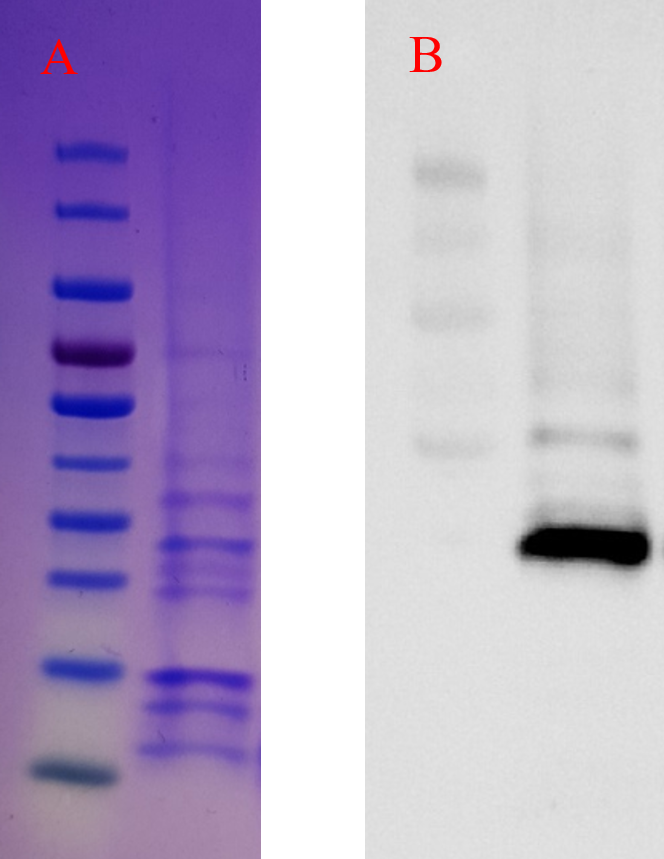
The BL21 samples induced with IPTG were run on Bio-Rad mini-protean TGX precast gel to evaluate the intracellular expression of the hEcad1/2 protein. As shown in Fig. 4A, a band of 23.7 kDa corresponding to the molecular weight of the hEcad1/2 was observed. For the periplasmic expression of hEcad1/2, we used pelB signal peptide, and as shown in Fig. 4B, Western blotting confirmed the expression of His-tagged protein.
3.3. Construction and cloning of recombinant p1CSV-CotY-N-hEcad1/2
The recombinant plasmid p1CSV-CotY-N-hEcad1/2 was constructed by inserting the hEcad1/2 DNA fragment into the p1CSV-CotY-N backbone vector (Fig. 2). Obtained colonies had an antibiotic selection, and the gene amplification from the construct showed the expected size. We confirmed no DNA mutation in the constructed plasmid by DNA sequencing (data not shown) and then transformed p1CSV-CotY-N-hEcad1/2 into competent B. subtilis KO7 cells.
3.4. Identification of cotY-hEcad1/2 integrated into the chromosome of B. subtilis
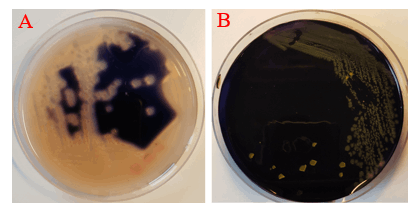
Amylase activity assay was employed to identify cotY-hEcad1/2 integration mutants. The integration of cotY-hEcad1/2 at amyE locus disrupted the secretion of amylase; as a result, no colorless halo was observed around the colonies on the Starch Agar plates stained by iodine, but in control strain, a white halo appeared around the colonies due to the secretion of amylase (Fig. 5).
3.5. Detection of L. monocytogenes
The reliability of the developed sporobeads for the detection of pathogenic L. monocytogenes was evaluated under the Zeiss Axioskope 2 plus microscope. As illustrated in Fig. 6, the sporobeads showed high-affinity binding to the L. monocytogenes 4b.

4. Conclusion
In this study, we aimed to develop a cost-effective and efficient method for detecting and capturing pathogenic L. monocytogenes. We utilized the adhesive properties of human E-cadherin domains 1 and 2 (hEcad1/2) for this purpose. Our approach involved the expression of hEcad1/2 on the spore coat of Bacillus subtilis, creating sporobeads capable of capturing L. monocytogenes. Our results demonstrated the successful expression and purification of hEcad1/2, which was efficiently incorporated into the spore coat of B. subtilis. The sporobeads generated in this manner exhibited a high binding affinity for L. monocytogenes 4b. This outcome suggests the potential of hEcad1/2 in practical applications involving the detection and capture of pathogenic L. monocytogenes. This method offers advantages in terms of cost-effectiveness and ease of production compared to traditional techniques. Furthermore, the specificity of hEcad1/2 for L. monocytogenes highlights its suitability for pathogen identification. In conclusion, our study provides a promising platform for the rapid detection and capturing of pathogenic L. monocytogenes using sporobeads coated with hEcad1/2. Further research and development in this direction could lead to valuable applications in food safety and clinical diagnostics.
References
[1] Pizarro-Cerdá J, Cossart P. Microbe profile: Listeria monocytogenes: a paradigm among intracellular bacterial pathogens. Microbiology. 2019 Jul;165(7):719-21. View Article
[2] Schubert WD, Urbanke C, Ziehm T, Beier V, Machner MP, Domann E, Wehland J, Chakraborty T, Heinz DW. Structure of internalin, a major invasion protein of Listeria monocytogenes, in complex with its human receptor E-cadherin. Cell. 2002 Dec 13;111(6):825-36. View Article
[3] Ireton K, Mortuza R, Gyanwali GC, Gianfelice A, Hussain M. Role of internalin proteins in the pathogenesis of Listeria monocytogenes. Molecular Microbiology. 2021 Dec;116(6):1407-19. View Article
[4] Harter E, Lassnig C, Wagner EM, Zaiser A, Wagner M, Rychli K. The novel internalins InlP1 and InlP4 and the internalin-like protein InlP3 enhance the pathogenicity of Listeria monocytogenes. Frontiers in Microbiology. 2019 Jul 23;10:1644. View Article
[5] Lecuit M, Ohayon H, Braun L, Mengaud J, Cossart P. Internalin of Listeria monocytogenes with an intact leucine-rich repeat region is sufficient to promote internalization. Infection and immunity. 1997 Dec;65(12):5309-19. View Article
[6] Luo Y, Lee A, Shen H, Radice GL. Altering tissue tropism of Listeria monocytogenes by ectopically expressing human E-cadherin in transgenic mice. Microbial pathogenesis. 2003 Aug 1;35(2):57-62. View Article
[7] Hibi K, Abe A, Ohashi E, Mitsubayashi K, Ushio H, Hayashi T, Ren H, Endo H. Combination of immunomagnetic separation with flow cytometry for detection of Listeria monocytogenes. Analytica Chimica Acta. 2006 Jul 28;573:158-63. View Article
[8] Harter E, Lassnig C, Wagner EM, Zaiser A, Wagner M, Rychli K. The novel internalins InlP1 and InlP4 and the internalin-like protein InlP3 enhance the pathogenicity of Listeria monocytogenes. Frontiers in Microbiology. 2019 Jul 23;10:1644. View Article
[9] Lecuit M, Hurme R, Pizarro-Cerdá J, Ohayon H, Geiger B, Cossart P. A role for α-and β-catenins in bacterial uptake. Proceedings of the National Academy of Sciences. 2000 Aug 29;97(18):10008-13. View Article
[10] Höfler C, Heckmann J, Fritsch A, Popp P, Gebhard S, Fritz G, Mascher T. Cannibalism stress response in Bacillus subtilis. Microbiology. 2016 Jan;162(1):164-76. View Article
[11] Russell AD. Bacterial spores and chemical sporicidal agents. Clinical microbiology reviews. 1990 Apr;3(2):99-119. View Article